Vervielfältigung von DNA durch die Polymerase-Kettenreaktion (PCR)
Hier kommt uns nun eine Technik zu Gute, die Mitte der achtziger Jahre entwickelt wurde und die als Polymerase Kettenreaktion (engl. Polymerase Chain Reaction, kurz PCR) bezeichnet wird. Dieses Verfahren erlaubt es, DNA-Abschnitte von Interesse aus einem Gesamtgemisch von DNA gezielt in einem solchen Grade zu vervielfältigen, dass sie letztendlich den Hauptanteil in dem Gemisch ausmacht und die restliche DNA, die uns nicht interessiert, für die Analyse nicht mehr störend ist.
Stellen wir uns einmal eine bunte Sommerwiese vor, auf der eine große Zahl unterschiedlicher Pflanzen wachsen. Wir sind nun speziell daran interessiert sagen wir mal einen Ackergauchheil zu finden. Würden wir nun ein Areal von einem Quadratmeter der Wiese untersuchen, so fänden wir vielleicht einen Löwenzahn, einen Hahnenfuß, ein paar Gänseblümchen, einige Standardgräser, aber nur mit viel Glück einen Ackergauchheil. Hätten wir aber eine Methode, den Ackergauchheil zur schnellen Vermehrung anzuregen, indem wir vielleicht eine Substanz, die das bewerkstelligen kann, über der Wiese versprühen, dann hätten wir möglicherweise eine Woche später in dem selben Areal 100 Ackergauchheile, neben denen die anderen Pflanzen unterrepräsentiert wären.
In einer vergleichbaren, allerdings rein biochemischen Weise funktioniert die PCR-Reaktion. Diese macht sich Biokatalysatoren, bestimmte Enzyme, zu Nutze, die eine Rolle bei der Vervielfältigung von DNA spielen. Eine Vervielfältigung von DNA ist unter verschiedensten physiologischen Zuständen von Bedeutung, insbesondere bei der Zellteilung, wo das Erbgut in gleicher Menge an die entstehenden Tochterzellen weitergegeben werden muss. Dies wird von einer Gruppe von Enzymen bewerkstelligt, die als DNA-Polymerasen bezeichnet werden. Sie sind dazu imstande, zu einem gegebenen DNA-Einzelstrang (wir erinnern uns, dass DNA in der Regel als gepaarter Doppelstrang vorliegt) den passenden komplementären (Negativ-) Strang herzustellen. Dazu benötigen die DNA-Polymerasen allerdings in der Regel eine kurze doppelsträngige Region als Startpunkt, von der aus sie ihre Synthese beginnen können.
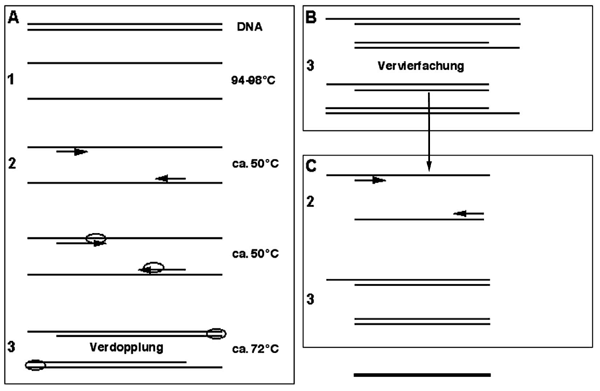
Im Reaktionsgefäß kann man dies dadurch erreichen, indem man die DNA-Doppelstränge durch Hitzeeinwirkung (94-98 Grad Celsius) trennt (siehe oben), um anschließend bei niedrigeren Temperaturen an bestimmten Stellen, die das uns interessierende Fragment flankieren, die Basenpaarung mit ca. 20-30 Nukleotiden langen sogenannten Oligonukleotiden zu ermöglichen. Speziell in der PCR-Anwendung werden diese Oligonukleotide als „Primer“ (sprich „Praimer“) bezeichnet. Diese Primer lagern sich, oder wie man sagt „hybridisieren mit“, (an) solche(n) Regionen (an), die in der Nukleotidabfolge komplementär zu ihnen sind und bilden dadurch kurze doppelsträngige Bereiche. Damit hat man nun durch gezielte chemische Synthese der Primer die Möglichkeit, den Startpunkt für die Vervielfältigung der gewünschten DNA-Region exakt festzulegen. In Gegenwart von einzubauenden Nukleotiden und einer aus bestimmten Organismen gewonnenen hitzestabilen DNA-Polymerase wird dann bei wiederum erhöhter Temperatur komplementär zu einem DNA Strang der entsprechende gegenläufige aufgebaut – also zum Negativ das Positiv und zum Positiv das Negativ (Abb. 5).
Bleiben wir beim Beispiel der Fotografie, obwohl das – wie meistens – etwas hinkt, da es ein zweidimensionales Gebilde darstellt und die DNA ihre Information in einer eher eindimensionalen Form enthält. Nehmen wir an, wir hätten eine riesengroße Fotografie eines Menschen und wir wollten nur die Region zwischen Nase und Mund genauer unter die Lupe nehmen und deswegen vervielfältigen. Dann würde die DNA-Polymerase einer Vervielfältigungsmaschine entsprechen, die allerdings erst noch wissen muss, welchen Bereich sie vervielfältigen soll. Dazu geben wir einen kleinen Teil des Bildes (den Primer), der als Negativ auf das Positiv in der Nasenregion passt, in das Reaktionsgemisch. Dieses Fragment (der Primer) kann außerdem so hergestellt werden, dass er auch die Richtung der Vervielfältigung – also hier in Richtung Mund – vorgibt. Gleichzeitig geben wir der Vervielfältigungsmaschine ein Fragment (wieder einen Primer) vor, das einem Positiv entspricht, welches auf das Negativ der Region Mund passt und in Richtung Nase zeigt. Die Maschine weiß nun „OK, ich muss Negativ und Positiv von Nase bis Mund vervielfältigen“. Das Schöne dabei ist, dass jeder neu entstandene Bildabschnitt aus der Region Nase bis Mund wieder als Vorlage für weitere Vervielfältigung dienen kann, was letztendlich eine exponentielle Vermehrung erlaubt. Je Vervielfältigungsdurchgang wird also verdoppelt, was theoretisch bei 40 Durchgängen schon zu einer Vervielfältigung um den Faktor eine Billiarde führt. In der Praxis ist die Effizienz allerdings wesentlich geringer.
Natürlich vervielfältigen wir bei der PCR-Reaktion auch nicht Nase bis Mund sondern Teile eines Genoms, die für weiß Gott was codieren oder auch nicht, aber ich hoffe, das Prinzip ist klar geworden. Mit Hilfe dieses Verfahrens können wir jedenfalls im Reaktionsgefäß mit Hilfe eines so genannten „Thermocyclers“, der in vorgegebenen Zeitabständen gewünschte Temperaturen einstellen kann, schon aus Spuren von DNA analysierbare Mengen von mehreren hundert Nanogramm fabrizieren. Dabei wählen wir die Primer immer so, dass sie sich an Regionen konservierter, also hoch erhaltener Sequenz heften können, welche die variablen Sequenzen flankieren.
Haben wir nun die Fragmente von Interesse in genügend hohem Maße vervielfältigt, so können sie auf prinzipiell zweierlei Art analysiert werden. Erstens können wir mit Hilfe moderner Sequenzierverfahren, die hier nicht erläutert werden sollen (automatische Fluoreszenzsequenzierung) die Nukleotidabfolge (eben die Sequenz) bestimmen. Diese variiert meistens für bestimmte Gene oder auch anderer Abschnitte des Genoms in typischer Weise zwischen verschiedenen Spezies. Haben wir also ausgehend von einer Gewebeprobe eines bestimmten Tieres die Sequenzen geeigneter Abschnitte einiger Gene bestimmt, so können wir meistens sehr schnell sagen, von welcher Spezies die Probe stammt. Darüberhinaus sind anspruchsvolle Sequenzvergleichsprogramme (Stichwort „Bioinformatik“) dazu imstande, anhand des Grades der Sequenzunterschiede untrerschiedlicher Genabschnitte auch auf den Verwandtschaftsgrad verschiedener Spezies in Hinblick auf ihre Evolution zu schließen.
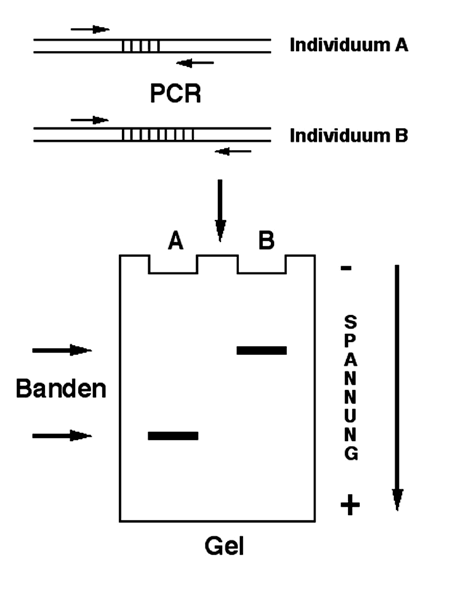 Abb. 6: Prinzip einer Mini/Mikrosatelliten- oder VNTR-Analyse.[/caption]
Abb. 6: Prinzip einer Mini/Mikrosatelliten- oder VNTR-Analyse.[/caption]Geht es – meist bei der Identifikation von Individuen – allein um die Unterschiede in dem Grad der Wiederholungen bestimmter Nukleotidabfolgen (Minisatelliten, Mikrosatelliten, VNTRs), die sich natürlich auf die Länge des durch PCR vervielfältigten Fragmentes auswirken, so können diese Längenunterschiede mit Hilfe der so genannten Gelelektrophorese sichtbar gemacht werden. Dies kann man sich folgendermaßen vorstellen: Man fertigt mit Hilfe bestimmter Apparaturen und Substanzen ein dünnes Gel, sagen wir mal 10 x 10 x 0,5 cm an, das im Grunde genommen nichts anderes als eine Art Pudding ist. Dieser Pudding hat an einer Seite „Taschen“, in die man die aus der PCR Reaktion hervorgegangenen DNA Proben hineinpipettieren kann. Dies geschieht natürlich auch in einer bestimmten Apparatur, in der sich das Gel unter einer definierten Flüssigkeit, einem „Puffer“, mit geeigneter Konzentration an Salzen und anderen Chemikalien befindet. Danach wird eine elektrische Spannung von einem Ende des Gels zum anderen angelegt. DNA trägt negative elektrische Ladungen und wandert daher – Gegensätze ziehen sich ja bekanntlich an – zum positiven Pol, der „Anode“. Dabei hat sie sich allerdings durch den Urwald vernetzter Makromoleküle zu kämpfen, der einen „Pudding“ ausmacht. Umso länger das DNA Molekül nun ist, um so effektiver wird es dabei gebremst werden, sich durch das molekulare Maschenwerk des Gels zu bewegen. Kleine DNA-Moleküle können die Freiräume zwischen den Gelmolekülen hingegen besser nutzen und wandern schneller. Nach einer gewissen Dauer der Gelelektrophorese werden kleinere DNA-Moleküle also deutlich weiter gewandert sein als die Großen, wobei Moleküle gleicher Größe auch immer gleich weit gewandert sind. Die im Gel befindlichen DNA-Moleküle werden dann mit Hilfe einer bestimmten Substanz angefärbt, welche unter ultravioletter Bestrahlung die DNA als fluoreszierende kurze Linien, die so genannten „Banden“, sichtbar macht. Anhand der Identitäten bzw. Unterschiede in den Wanderstrecken der vervielfältigten DNA-Fragmente aus unterschiedlichen Gewebeproben kann man dann auch auf die Identitäten verschiedener Individuen schließen (Abb. 6). Sehr kompliziert – ich weiß – aber es funktioniert. Dieses Verfahren hat in der Kriminalistik schließlich ausgehend von nur geringen Zellresten, die an Zigarettenkippen hängen, oder Spermaresten schon oft erfolgreich zur Identifikation von Tätern geführt.
Ein Kuriosum, dass manche LeserInnen bestimmt zum Schmunzeln bringen wird, soll hier noch erwähnt werden: Das CITES Abkommen verbietet den internationalen Handel mit bedrohten Tieren und deren Bestandteilen. Darunter zählen in der Tat auch DNA-Proben. Bestimmte PCR-Techniken erlauben jedoch die gezielte Isolierung der während der PCR-Reaktion durch Vervielfältigung neu aufgebauten DNA-Stränge, die nicht mehr als ein ursprünglicher Bestandteil des zu analysierenden Tieres angesehen werden. Diese Proben dürfen dann auch über Landesgrenzen transportiert werden. Ein nützlicher Schachzug, der erst durch die PCR-Technik ermöglicht wurde.
